基于分子动力学模拟的静电场下LiCl溶液的水化结构
东南大学能源与环境学院 严攀 殷勇高
【摘 要】LiCl吸湿溶液由于其节能、可循环利用等优点在空气调节领域得到了广泛应用,同时电场被多次证实可以用于强化气液传质,但除湿及电场强化的机理尚不明确。为了探究LiCl溶液吸湿的过程及为研究静电场强化传质效果的机理分析打下基础,本文采用分子动力学探究静电场对LiCl溶液的水化结构的影响,首先选取了常用的三种的LiCl水溶液的力场,并根据模拟物性的能力选出最适合LiCl的力场即Koneshan模型。因此本文用Koneshan模型研究静电场对溶液体系的水化结构和影响传质的表面张力的影响,发现垂直于气液界面的电场会使水分子倾向于定向排列,从而降低了LiCl的表面张力,但静电场并不影响离子-水分子的水合结构。
【关键词】分子动力学模拟 溶液除湿 电场强化 水合结构
【中图分类号】TU831.6
【基金项目】国家自然科学基金自助项目(52076039)、国家重点研发计划课题资助项目(2018YFC0705306)。
Abstract: LiCl solution has been widely used in the field of air conditioning due to its energy-saving and recyclability, and electric fields have been repeatedly proved to be able to enhance gas-liquid mass transfer, but the mechanism of dehumidification and electric field enhancement is unclear. To explore the moisture absorption process of LiCl solution and lay the foundation for studying the enhancement mechanism of electrostatic field, molecular dynamics simulation is adopted to explore the effect of electrostatic field on the hydration structure of LiCl solution. Three different force fields of LiCl are explored firstly, and the most suitable one is selected according the ability of reproducing physical properties. Therefore, this paper uses model of Koneshan to study the effect of electrostatic field on the Hydration structure and surface tension of the solution system. It is found that the electrostatic field perpendicular to the gas-liquid interface tends to align the water molecules, thus reducing the surface tension of LiCl, but the electric field does not affect the hydration structure of ion-water molecules.
Keywords: molecular dynamics simulation; solution dehumidification; electric field enhancement; hydration structure
卤盐溶液因其吸湿性、节能性、易实现、可循环利用的特点在空气调节领域得到了广泛应用[1,2],其中LiCl溶液由于其除湿性能优越得到了广泛的关注。但目前的研究手段主要停留在宏观实验层面和经验关联式的拟合[3],缺少对分子尺度的探究。同时有研究表明,外部的物理场强可以用来强化气液传质,其中静电场可以通过极化效应对物系产生影响,且易于控制和调节,已经成为近年来化工过程强化领域研究和开发的热点[4],静电场对溶液除湿/再生的影响及机理鲜有报道,了解除湿/再生过程及其强化过程的微观机理对于拓宽溶液除湿这一技术的应用和理论发展有重要意义。
分子动力学模拟(MD)正逐渐成为多种学科范围内的有效研究手段,已经广泛应用于模拟生物蛋白、蒸发、冷凝、气液界面等现象[5,6]。对于模拟最重要的是选取能够描述粒子间作用力的势能函数参数即力场,针对电解质溶液的力场主要包括非极化力场[7-9]和极化力场[10,11]。LiCl溶液中包括水氢原子、氧原子以及Li+和Cl-,而离子的力场模型多数是在水模型的基础上发展而来,通过拟合水合自由能、离子水合半径、径向分布函数、晶格能等来获取参数,力场参数是主要是离子的势能阱ε、软球直径σ以及极化率。Jorgensen[7]等报道了金属离子F-,Cl-,Br-,以及I-和一些碱性金属离子的Lennard Jones参数,这些参数是结合TIP4P的水分子模型得出的,同时考虑了不同边界条件的影响,为了拟合出正确的水化能以及第一壳层的径向分布函数,同时复现出了和从头算得出的结果相同的一水合物的能量而优化出的结果,这是第一个完整且自洽的用于模拟的离子参数集。Joung[8]等为了复现离子的一系列性质,包括结构、运动、溶解、结晶态、以及和其他离子相互作用方面的参数,采用集中常用的水分子模型以及Ewald加和的算法,首次在优化σ和ε时把平衡阴阳离子的晶格能LE和晶格常数LC参数纳入拟合的考虑范围内。Koneshan等[9]使用25°C的SPC/E模型和长程作用的反应场,考虑了水化结构以及滞留时间等因素,结果显示计算结果较好。极化模型应用较多的是基于SWM4-NDP水模型的Drude模型,其中的离子参数由Yu[12]拟合得出,但目前只在低浓度中适用良好。综合来看,非极化力场计算量较小,且被多次证实可以准确地预测溶液的多种热力学性质[13]。水合结构不仅仅是发展力场的重要考虑因素,也是水和溶液存在诸多性质差异的根本原因。在电解质溶液中,离子附近形成的电场使部分溶剂水分子在离子周围发生定向排列,离子水合层中水的微观结构发生改变,这就是溶液中离子对水结构的影响,而电场的加入则会影响溶液本身的内部电场从而影响水化结构及水分子偶极取向[14],带来宏观物性表面张力的改变,同时表面张力是影响传质的重要因素,因此对电场下溶液水化结构开展分子动力学模拟有重要意义。
从上述综述中可以看出,针对电解质溶液的力场有多种参数,而适用于LiCl溶液的力场参数并没有最佳选择,这也阻碍了后续的盐溶液除湿过程模拟以及强化机理分析的脚步。考虑到模拟过程的巨大计算量和模拟时间,本文优先选择计算效率较高的非极化力场,通过比较物理性质的实验值和分子动力学模拟值来选择适合LiCl溶液的力场,并应用所选择力场模拟LiCl溶液的水化结构和对传质影响较大的表面张力,对比研究有无静电场下溶液体系的变化,为从分子视角研究盐溶液除湿过程和电场强化盐溶液-空气体系热质交换的机理奠定基础。
1 方法
本文的模拟对象为LiCl水溶液,采用开源软件GROMACS进行300-340K系列温度条件下的LiCl溶液的物性和结构模拟,模拟体系如图1所示,共由596个水分子、141个Li+和141个Cl-组成,针对密度模拟、比热容等模拟时采用模型a(2.9×2.9×2.9nm),模拟表面张力时采用模型b(2.9×2.9×8nm)。对于分子数目的限制可能会导致小样本系统的表面效应掩盖其本体效应,不能完全反应真实系统的性质和行为,因此在本课题研究中,采取周期性边界条件的方法模拟浓度为35%的LiCl溶液。在模拟过程中,只考虑分子之间非键作用力,包括范德华力Evdw、静电库伦力Eelec(包括了氢键)。因此,总的势能计算如下:
Etotal=Evdw+Eelec (1)
范德华力和经典库仑力的计算式如下:
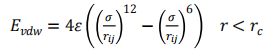 (2)
(2)
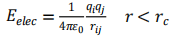 (3)
(3)
LJ势中的离子的势能阱ε以及软球直径σ以及电荷量参数q列于表1,分别是来自Jorgensen、Joung和Koneshan的拟合数据[7-9]。粒子之间作用力的计算方式可以分为几何混合法则和算术混合法则[15],GROMACS已实现通过Settle算法对刚性水分子进行约束,为了模拟过程的不崩溃首先需要对整个系统进行能量最小化以达到势能最小的构型,并在NPT系综下进行温度和压力的预平衡,压力在过程中始终控制在1atm,控制温度的算法采用Velocity-rescaling算法,控制压力的算法采用Parrinello-Rahman,处理长程静电相互作用力采用PME的方法,为了减少计算量,使用1nm的截断距离,时间步长取为2fs。所有的过程采用稳态模拟,分为预平衡阶段和数据采集阶段。预平衡过程使用2ns来确保温度和压力达到稳定,并用额外的1ns每隔1ps来采集数据,计算密度、定压比热容及扩散系数。
为了模拟计算表面张力,需要营造一个气液界面,将模拟盒子的高度拉伸至8nm,模拟体系在三个方向都采用周期边界条件。模型的高度计得较长以保证足够的汽相空间。液膜设计得足够厚以防止液膜两侧的分子相互作用,模拟时间和步长与前述相同。当考虑静电场后,在GROMACS中设定不同大小的恒定电场以进行控制变量法比较,如图1(c)所示,在体系的Z轴方向施加0-2 V/nm的电场,如图6所示。为了防止电场的引入使模拟坍塌,需要进行时间更长的预平衡,其余计算参数不变。
表1 选取的力场参数
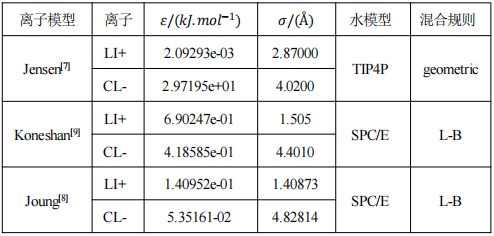
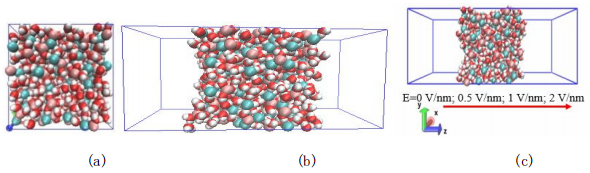
图1 模型示意图(其中白色为H,红色为O,粉色为Li,绿色为Cl)
2 力场评价结果和讨论
35%浓度的LiCl水溶液的密度如图2所示,温度范围为300K至340K,总体而言,三种模型都能够模拟出密度随温度变化的趋势。与文献中的实验值相比,所有模型都可以再现LiCl水溶液的密度[16]。 Jensen等人的模型给出最佳的密度估计,平均相对误差为4.0%,其次是Koneshan等人的模型,最大相对误差为4.4%。图3显示了不同温度的LiCl水溶液中水分子的扩散系数,在300K的温度下,Jensen模型给出的预测值约为1.26×10-5 cm2/s,根据文献实验[17],在相同工况下水在LiCl溶液中的扩散系数约为1.0*10-5 cm2/s,总体上Jensen模型更加接近扩散系数的真实值。根据涨落理论算出定压比热容Cp,如图4所示。将模拟的定压比热容与文献中的实验值[16]相比,三种模型的表现都有较大误差,其中复现能力最差的是Joung模型,Jensen和Koneshan模型的准确程度相似。LiCl水溶液在300 K、320K、340 K时的表面张力模拟值如图5所示,所有的模型都能正确地模拟出表面张力随温度的变化趋势。由于Jensen对表面张力的模拟误差过大,只在此处比较Joung和Koneshan模型的准确性,与实验值[18]相比, Koneshan力场的模拟结果优于Joung的结果,Koneshan力场的平均相对误差为8%,而Joung力场的平均相对误差为10.3%。
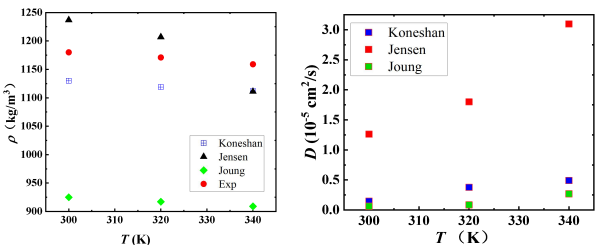
图2 LiCl溶液密度模拟值 图3 水在溶液中的扩散系数
根据密度,比热容,扩散系数和表面张力的模拟结果,Jensen等人提出的力场能够以最高的精度再现实验密度和扩散系数,但模拟表面张力表现最差。Joung等人提出的力场在物性的模拟方面没有复现比较好的值。与此相反的是Koneshan等人提出的力场,不仅能够产生与实验数据相比最佳的表面张力,还具有令人满意的其他热力学性质复现的能力。这表明了Koneshan等人的模型优于其他两者。因此,本文将使用Koneshan力场来进行分子动力学模拟。
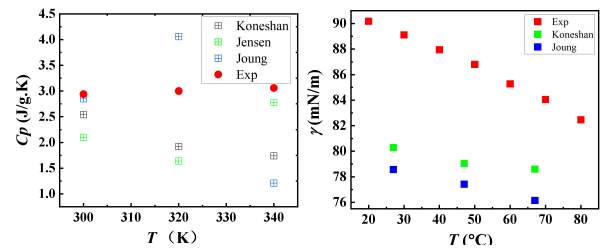
图4 LiCl的定压比热容 图5 LiCl溶液的表面张力
3 静电场对LiCl溶液水化结构影响
解质溶液中阴离子和阳离子与水分子之间存在着强烈的库仑相互作用,形成了较大的溶剂化壳层。未加电场时的溶剂-离子结构可以从图6的径向分布函数中得出。径向分布函数(radial distribution function, RDF)的物理意义可由图6显示,图中,中心原子为“参考分子”,与其中心的距离由r→r+dr间的分子数目为dN。定义径向分布函数g(r)为:
ρg(r)4πr2=dN (4)
式中,ρ为系统密度。径向分布函数(RDF)可以解释为系统的区域密度与平均密度的比值,参考分子的附近区域密度不同于系统的平均密度,但与参考分子的距离较远时应与平均密度相同,即当r值大时径向分布函数函数的值应该接近1。
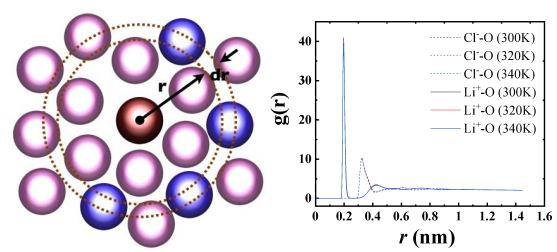
图6 径向分布示意 图7 Li+、Cl-相对于氧原子的径向分布函数
图7显示了阴阳离子和氧原子之间的径向分布函数,Li+与氧原子的径向分布函数在0.196 nm 处有一个强烈的峰值;Cl-与氧原子的径向分布函数在0.32nm处有一个峰值,该处为第一水合层,可以看出水分子中的氧原子与Li+更近,其原因是氧原子呈电负性,更容易和带正电的离子相结合,因此与氧原子与Li离子之间的相互作用力更强。从图6还可以得出在300-340K温度区间内,温度对径向分布函数的影响很小,这与纯水的情况不一样,在纯水中,温度会使O-O,O-H径向分布函数发生显著变化,其原因是高温带来的氢键的破坏[19]。
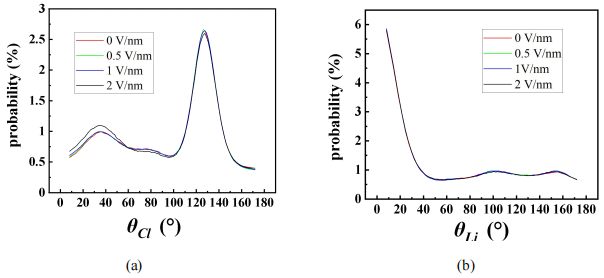
图8 离子周围水分子取向(其中a为Cl-周围水分子取向,b为Li+周围水分子取向)
图8给出了不同大小的电场施加下,Cl-和Li+周围水分子的取向角在[0°,180°]范围内的分布概率,横坐标为离子到氧原子的向量与水分子偶极之间的夹角,从取向角分布可以得出更具体的水化结构信息。从图8(a)中可以看出,在127.58° 时的概率有峰值,Cl-周围水分子取向角大于150° 或小于100° 的概率都很小。从图8(b)得出Li+周围水分子取向角在约8°时的概率有峰值,并且角度越大,Li+周围水分子取向角在该角度分布的概率越小。由上可见其O-H共价键指向阴离子,而氧原子与Li+较近,Li+接近位于水分子角平分线上。结合径向分布函数的规律可以描述出整体的水分子和离子的分布情况,如图9所示。随着电场强度的增加的增大,离子周围水分子取向角概率峰值位置不变,大小有轻微的改变。这就说明尽管水分子会受到电场的影响导致总体偶极矩的方向有所改变,但是由于水合作用带动离子随溶剂一起转动从而保证了相对固定的水合结构。
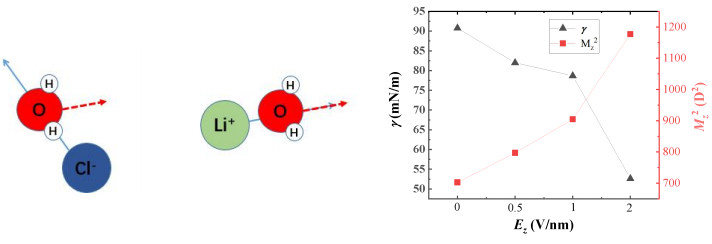
图9 离子周围水分子的排布倾向 图10 电场对表面张力及总偶极矩的影响
图10给出了不同大小的电场对表面张力的影响。能够得出对于平行电场而言,方向垂直于气液界面的电场使表面张力减小,这可以归因于垂直的电场可以使水分子这种极性分子趋向于定向排列[14],从而减小了粒子之间的作用力,表现为表面张力减小,定向排列的现象也可由图8中Z轴方向的总偶极矩分量Mz的变化趋势推出,由于模拟过程中采用的刚性水分子,所以每个水分子的极矩为固定的2.35D,但增大的总的Z轴偶极矩说明了水分子在Z轴方向电场的施加下倾向于Z轴的定向排列。
4 结论
为了从微观角度探究LiCl吸湿溶液及为研究静电场强化传质效果的机理分析打下基础,本文采用分子动力学方法,探索了描述LiCl水溶液中粒子相互作用的合适力场。根据密度、比热容、扩散系数、表面张力等模拟数据和实验值的对比情况,在三个最新的非极化力场中选择了Koneshan模型,进一步选模拟电场对溶液体系的微观结构、表面张力的影响,并进行了初步分析,主要结论如下:
(1)尽管非极化力场存在一定的缺陷,但由于其计算性能高且拥有复现热力学参数的良好能力,依然有很大的利用价值。Koneshan等人的模型给出了最佳的表面张力模拟,并显示出适中的预测其他性能的能力,这表明Koneshan等人的模型可以更好地用于包含气液界面的模拟。
(2)垂直于气液界面的电场会使LiCl的表面张力减小,且静电场会使水分子倾向于Z轴定向排列,但并不影响离子-水分子的水合结构,Li+比Cl-的水合半径更小,O-H共价键指向阴离子,Li+在偶极反方向的水分子角平分线上。
符 号 说 明
参考文献
[1] Yin Y G, Zhang X S. Comparative study on internally heated and adiabatic regenerators in liquid desiccant air conditioning system[J]. Building and Environment, 2010, 45(8): 1799-1807.
[2] Zhang F, Yin Y, Zhang X. Performance analysis of a novel liquid desiccant evaporative cooling fresh air conditioning system with solution recirculation[J]. Building & Environment, 2017, 117(MAY): 218-229.
[3] Asfand F, Stiriba Y, Bourouis M. CFD simulation to investigate heat and mass transfer processes in a membrane-based absorber for water-LiBr absorption cooling systems[J]. Energy, 2015, 91: 517-530.
[4] 马空军, 贾殿赠, 孙文磊, et al. 物理场强化化工过程的研究进展. 现代化工, 2009: 33-37,39.
[5] Mieda S. Analysis of the Interaction between a Protein and Polymer Membranes Using Steered Molecular Dynamics Simulation to Interpret the Fouling Behavior[J]. Bulletin of the Chemical Society of Japan, 2020, 93(12): 1443-1448.
[6] Xue L, Keblinski P, Phillpot S R, et al. Effect of liquid layering at the liquid-solid interface on thermal transport[J]. International Journal of Heat and Mass Transfer, 2004, 47(19-20): 4277-4284.
[7] Jensen K P, Jorgensen W L. Halide, Ammonium, and Alkali Metal Ion Parameters for Modeling Aqueous Solutions[J]. J Chem Theory Comput, 2006, 2(6): 1499-509.
[8] Joung I S, Cheatham Iii T E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations[J]. The journal of physical chemistry B, 2008, 112(30): 9020-9041.
[9] Koneshan S, Rasaiah J, Lynden-Bell R, et al. Solvent Structure, Dynamics, and Ion Mobility in Aqueous Solutions at 25 °C[J]. Journal of Physical Chemistry B, 1998, 102: 4193-4204.
[10] Dang L. Mechanism and Thermodynamics of Ion Selectivity in Aqueous Solutions of 18-Crown-6 Ether: A Molecular Dynamics Study[J]. Journal of The American Chemical Society - J AM CHEM SOC, 1995, 117.
[11] Dang L. Development of Nonadditive Intermolecular Potentials Using Molecular-Dynamics - Solvation of Li+ and F- Ions in Polarizable Water[J]. The Journal of Chemical Physics, 1992, 96: 6970-6977.
[12] Yu H, Whitfield T, Harder E, et al. Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field[J]. Journal of chemical theory and computation, 2010, 6: 774-786.
[13] Vega C, Abascal J L F. Simulating water with rigid non-polarizable models: a general perspective[J]. Physical Chemistry Chemical Physics, 2011, 13(44): 19663-19688.
[14] Wang B-B, Wang X-D, Duan Y-Y, et al. Molecular dynamics simulation on evaporation of water and aqueous droplets in the presence of electric field[J]. International Journal of Heat and Mass Transfer, 2014, 73: 533-541.
[15] Kong C L. Combining rules for intermolecular potential parameters. II. Rules for the Lennard‐Jones (12–6) potential and the Morse potential[J]. The Journal of Chemical Physics, 1973, 59(5): 2464-2467.
[16] Conde M R. Properties of aqueous solutions of lithium and calcium chlorides: formulations for use in air conditioning equipment design[J]. International Journal of Thermal Sciences, 2004, 43(4): 367-382.
[17] Potnis S V, Lenz T G, Dunlop E H. Measurement of water diffusivity in aqueous lithium bromide and lithium chloride solutions[J]. Chemical Engineering Communications, 1995, 139: 41-49.
[18] Wen Y, Bjurstrom H, Setterwall F. Surface-Tension of Lithium Bromide Solutions with Heat-Transfer Additives[J]. Journal of Chemical and Engineering Data, 1991, 36(1): 96-98.
[19] Shvab I. Molecular dynamics simulation of water and aqueous solutions[D]. Swinburne University of Technology, 2014.
备注:本文收录于《建筑环境与能源》2021年4月刊 总第42期(第二十届全国暖通空调模拟学术年会论文集)。版权归论文作者所有,任何形式转载请联系作者。